Opitz G/BBB (Opitz) syndrome is a hereditary disorder that affects people in different ways, causing malformations in medial (midline) organs and structures, intellectual disability and developmental disorders. Scientists have revealed a new control mechanism for the gene that causes this disorder, a discovery that could help in developing treatment for the syndrome. The findings were published on May 16 in the online edition of Development.
A group of scientists led by Associate Professor UEYAMA Takehiko and Professor SAITO Naoaki (both from the Kobe University Biosignal Research Center) and members of Kyoto Prefectural University of Medicine carried out this research.
Professor Ueyama expressed his hopes that this discovery would contribute to “revealing the underlying mechanism that explains the range of symptoms caused by Opitz syndrome, a disease that has different effects on individual patients, even within the same family”.
Opitz syndrome occurs for at least 1 in every 10,000-50,000 people. It is a hereditary disorder that causes a wide range of physical malformations in midline structures of organs, including in the brain, the face, the heart, the larynx and pharynx, the trachea and esophagus, urinary organs and genitals.
Previous findings identified Midline 1 (MID1) as the gene responsible for Opitz syndrome. The functional decline of MID1 causes the congenital disorders described above, but it is still unclear why these symptoms are so varied among individual patients. Treatment methods are yet to be fixed, and surgical therapy is currently the main treatment.
The research team focused on cerebellar granule neurons, a type of neurons with the largest population in the brain, and a signaling protein/molecule called Rac which functions in cerebellar granule neurons during cerebellar development. The team created a “knockout” mouse with the Rac protein deleted. They discovered that this mouse experienced severe walking impairment because of the loss of the internal granule layer in the medial cerebellum. Next, they extracted the cerebellar granule neurons affected by the deleted Rac from the medial cerebellum. Using DNA microarrays they examined these neurons and discovered reduced expression of MID1, the causative gene of Opitz syndrome. This showed that Rac had been regulating the expression of Mid1, and when Rac was deleted, MID1 stopped functioning correctly in the mouse.
They also discovered a cell signaling pathway in which Rac-Mid1-mTOR form a complex and contribute to the differentiation and maturation of cerebellar granule cells.
The individual variability in these cell signaling pathways could be a cause of the broad range in the symptoms caused by Opitz syndrome. These findings could lead to development of a new treatment for Opitz syndrome that targets cell signaling.
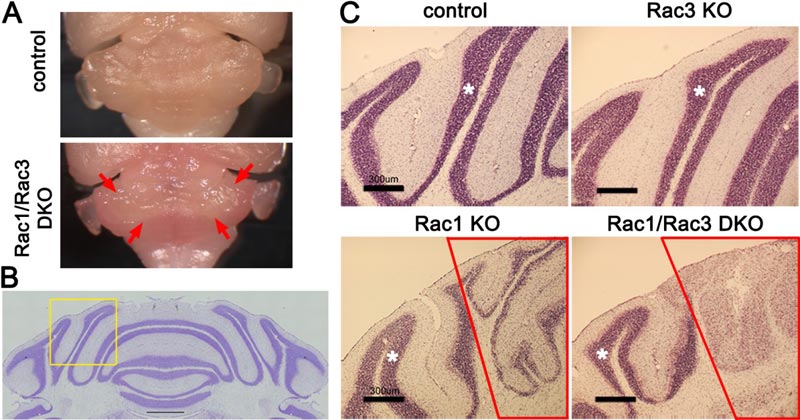
A: The Rac1/Rac3 double knockout (DKO) mouse cerebellum is smaller than the control specimen. The medial cerebellum surrounded by the arrows is the affected area – it is whiter than the areas surrounding it, which are pink.
B: A section of the control cerebellum stained with nissl solution. C shows enlarged parts of the nissl-stained areas in each mouse genotype in the area indicated by the yellow frame.
C: In the control and the Rac3 knockout (KO) mice the internal granule layer, stained a red-purple color, is healthy. On the other hand, the medial part of the internal granule layer (in the red frame) is thinner in the Rac1KO mouse and has vanished in the Rac1/Rac3 mouse. Scale bar: 300 μm
Journal information
- Title
- Novel role of Rac-Mid1 signaling in medial cerebellar development
- DOI
- 10.1242/dev.147900
- Authors
- Takashi Nakamura, Takehiko Ueyama, Yuzuru Ninoyu, Hirofumi Sakaguchi, Narantsog Choijookhuu, Yoshitaka Hishikawa, Hiroshi Kiyonari, Masaaki Kohta, Mizuho Sakahara, Ivan de Curtis, Eiji Kohmura, Yasuo Hisa, Atsu Aiba, Naoaki Saito
- Journal
- Development, 144 (10), 1863-1875, 2017