神戸大学バイオシグナル総合研究センターの上山健彦准教授と齋藤尚亮教授のグループ及び京都府立医科大学耳鼻咽喉科・頭頸部外科のグループらは、多発性で種々の正中部形成不全 (奇形) を呈する遺伝性疾患であるOpitz (オピッツ) 症候群の原因遺伝子 Midline 1 (Mid1) の新たな制御機構を発見しました。今後、Opitz症候群が呈する症状多様性の発症機序解明や新たな戦略に基づくOpitz症候群の治療法開発に貢献することが期待されます。
この研究成果は、5月16日 (日本時間) に、英国の発生生物学専門誌「Development」にオンライン掲載されました。
※ Development は、2009年に Special Libraries Association の BioMedical & Life Sciences Division (医学生物学部門) の Top 100 journals に選ばれています。
研究の背景
Opitz G/BBB症候群 (Opitz症候群、オピッツ症候群) は、出生率が1人/10,000~50,000で、脳 (小脳正中部低形成・脳梁欠損・発達遅滞)、顔面 (両眼解離・口唇口蓋裂)、心臓 (心房・心室中隔欠損)、喉咽頭・気管食道 (気管食道瘻)、泌尿・生殖器 (尿道下裂・鎖肛) など、多発性で種々の正中部形成不全 (奇形) を呈する遺伝性疾患である。
原因遺伝子としてMidline 1 (Mid1) が同定されており、その機能低下が上述の先天性異常 (奇形) を引き起こすことは解かっていたが、症状の多様性、即ち、個々の患者においてのみならず同一家系においてさえも症状が違う原因は未だ不明である。
また、治療法は確定しておらず、症状に対応した外科的療法が主である。
研究の内容
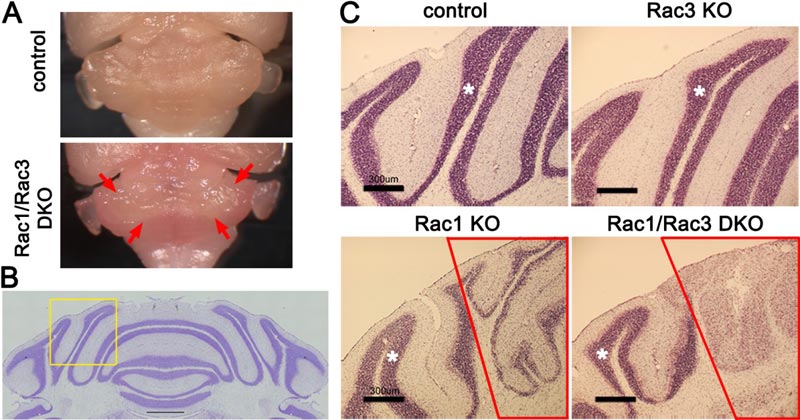
上山健彦准教授と齋藤尚亮教授のグループ及び京都府立医科大学耳鼻咽喉科・頭頸部外科のグループらは、Rhoファミリー低分子量G蛋白質であるRacを小脳顆粒細胞で特異的に欠損するノックアウトマウスを作製し、このマウスが小脳正中部の内顆粒層※2の欠損により高度の歩行障害を呈することを発見した。次に、このマウスからRacノックアウト小脳顆粒細胞を取り出し、DNAマイクロアレイ※3行ったところ、多発性で種々の正中部形成不全 (障害) を呈するOpitz症候群の原因遺伝子である Mid1 の発現が低下することを発見した。
更に、(1) Mid1の発現がRacにより転写※4レベルで制御される、(2) Rac-Mid1-mTOR※5が複合体となり小脳顆粒細胞の分化・成熟を促進する、という新規の細胞内シグナル経路を発見した。
今後の展開
Opitz症候群は、原因遺伝子として Mid1 が同定されているが、症状の多様性、即ち、個々の患者においてのみならず同一家系においてさえも症状に違いが生じる原因は依然不明のままであり、Mid1 に加えて他の因子やシグナル経路による制御が考えられていた。
今回、我々が発見した新しい細胞内シグナル経路 (RacによるMid1の発現制御とRac-Mid1-mTOR複合体による小脳顆粒細胞の分化・成熟誘導) の個体差が、Opitz症候群の症状多様性を規定している可能性がある。
Opitz症候群の治療法は確立しておらず、症状に対応した外科的療法が主である。今回の発見は、細胞内シグナルをターゲットとした新たな戦略に基づくOpitz症候群の新規の治療法開発に繋がる可能性がある。

用語解説
- ※1 低分子量G蛋白質
- 特異的な標的分子に結合して細胞内シグナルを伝達する分子スイッチとして機能する。その中でRhoファミリーは20種類の分子が存在し、細胞の分化・形態・運動に機能することが知られている。
- ※2 内顆粒層
- 小脳顆粒細胞により形成される小脳皮質 (表層) の第3層。
- ※3 DNAマイクロアレイ
- 細胞内で発現している遺伝子情報を網羅的に検出する手法。
- ※4 転写
- DNAからRNAを合成する段階。RNAから蛋白質が合成される段階は翻訳。
- ※5 mTOR
- 細胞内シグナル伝達に関与するタンパク質リン酸化酵素の一種。
論文情報
- タイトル
- Novel role of Rac-Mid1 signaling in medial cerebellar development
- DOI
- 10.1242/dev.147900
- 著者
- Takashi Nakamura, Takehiko Ueyama, Yuzuru Ninoyu, Hirofumi Sakaguchi, Narantsog Choijookhuu, Yoshitaka Hishikawa, Hiroshi Kiyonari, Masaaki Kohta, Mizuho Sakahara, Ivan de Curtis, Eiji Kohmura, Yasuo Hisa, Atsu Aiba, Naoaki Saito
- 掲載誌
- Development, 144 (10), 1863-1875, 2017