神戸大学大学院工学研究科の小野倫也教授、植本光治助教、大学院生の小松直貴さんらと、北海道大学の江上喜幸助教らの研究グループは、第一原理計算に基づいたSiC中の高密度窒素層の構造モデルの提案を行いました。この成果は、SiC半導体デバイスの製造時の窒素アニールの働きについて理解を深めることにつながります。
この研究成果は、11月30日に Journal of the Physical Society of Japan に掲載されました。
ポイント
- 次世代のパワー用途向け半導体材料であるSiC (シリコンカーバイド) によるデバイスの性能向上には窒素系ガスをもちいた熱処理 (窒素アニール) が不可欠とされている。
- 窒素アニールによってSiC中に侵入した窒素原子が原子スケールでどのような構造を取るかはこれまでよく分かっていなかった。
- 今回、研究チームはコンピュータシミュレーションを駆使して、SiC中に形成される高密度の窒素原子層の構造を理論的に予測し、実験と整合する結果が得られることを確認した。
- これらの情報はSiC半導体素子の製造技術の改善に役立つと期待される。
研究の背景
SiC (シリコンカーバイド) は炭素とシリコンの化合物からなる半導体材料で、大電力制御を目的としたパワーエレクトロニクス用途向けの次世代半導体材料として注目を集めています。とくに「MOSFET」(Metal-Oxide-Semiconductor Field-Effect Transistor: 金属酸化膜半導体電界効果トランジスタ) とよばれるデバイスにSiC半導体を用いることで、SiCのもつ高耐圧性・高速応答性などの優れた特性を活かした、従来のシリコン半導体を凌駕する高性能な電力変換デバイスが実現できると期待されています。
しかしながら、現在のSiC-MOSFETではキャリア移動度とよばれる特性値の改善が大きな課題として残されています。MOSFETの製造では、SiC表面上にSiO2 (二酸化ケイ素) の絶縁被膜の形成 (「酸化処理」) が必要になりますが、処理中に偶発的に入り込む欠陥によりキャリア移動度が著しく低下することが知られており、このため長らくSiC-MOSFETデバイスは期待された性能を発揮することが難しい状況にありました。
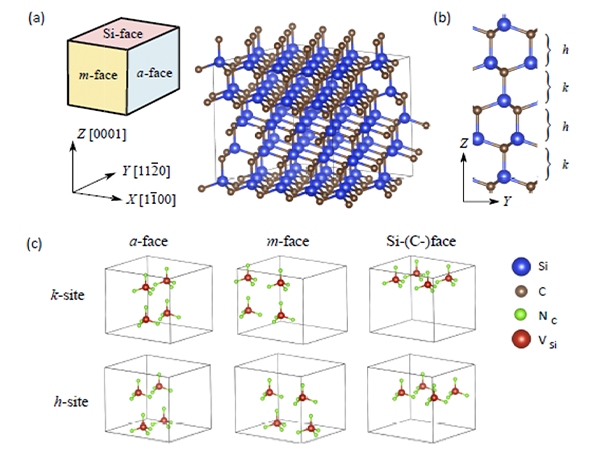
近年では、窒素系ガス中での熱処理 (窒素アニール) により、SiC-MOSFETの特性が向上することが報告されています。実験によると表面には高密度 (1平方cmあたり1014~1015個) の窒素原子が層状に存在すると考えられます。SiC結晶は特性の異なるさまざまな結晶面をもちますが[図1(a) 参照]、窒化のしやすさは面方位にも依存することが知られています。このように、様々な結晶面の方位を考慮したとき、原子スケールで見たときに窒素原子がどのような構造を取るのかよく理解されてきませんでした。
研究の内容
本研究では、SiC結晶中に形成される高密度窒素層の理論モデルを提案しました。本モデルはさまざまな結晶面の方位 (面方位) に対する窒素原子の配置を普遍的に記述することができます。提案された構造は4H-SiCのバルク結晶中に窒素原子 (NC) とそれを取り囲むようなシリコン空孔 (VSi) を添加したもので、不対結合を生じないため化学的安定性が予想されます。また、窒素原子密度は一平方センチメートル辺り1.2~1.5×1015個程度のとなり、実験で報告されている数値をよく再現できています。
さらに、当研究グループで開発を進めている第一原理電子状態計算プログラム「RSPACE」を用いたシミュレーションにより、構造最適化とエネルギーの安定性評価を行いました。形成エネルギーの面方位依存性から窒素添加の起こりやすさには異方性があり、今回のバルク結晶の場合はa面に沿った窒化が優位になることを理論的に予測しました。さらに詳細な解析を進めたところ、この挙動が窒素原子近傍のシリコン原子の窒素・炭素配位数に由来した電子状態の変化に起因することが明らかになりました。窒化の異方性は先行する実験からも報告されていましたが、原子スケールからみたメカニズムの解明は本研究が初の試みとなります。
今後の展開
SiC-MOSFETの高性能化には窒化処理は不可欠ですが、界面で起きる化学反応や窒素の侵入深さなど未解明な点は多く、今後、コンピュータによるシミュレーションを駆使した解析研究が進んでいくと考えられます。本研究で提案された高密度窒素層の構造モデルは、窒化された界面をコンピュータで扱うための低コストな計算モデル構築に役立つと期待しています。
謝辞
本研究は、文部科学省「富岳」成果創出加速プログラム「省エネルギー次世代半導体デバイス開発のための量子論マルチシミュレーション」(JPMXP1020200205) の一環として実施されたものです。また、本研究の一部は、スーパーコンピュータ「富岳」の計算資源の提供を受け、実施しました (課題番号: hp200122/hp150273)。また、本研究成果の一部はJSPS科学研究費 (JP16H03865) の助成のもと行われ、計算資源として東京大学物性研究所の全国共同利用によるSystem-BおよびSystem-C、筑波大学計算科学研究センター学際共同利用プロジェクトによるスーパーコンピュータOakforest-PACSも使用されました。
論文情報
- タイトル
- “First-Principles Study on Structure and Anisotropy of High N-atom Density Layer in 4H-SiC”
- DOI
- 10.7566/JPSJ.90.124713
- 著者
- Mitsuharu Uemoto, Naoki Komatsu, Yoshiyuki Egami and Tomoya Ono
- 掲載誌
- Journal of the Physical Society of Japan